Sintesi, nomenclatura e proprietà del gruppo funzionale delle ammidi
In questo post, cercheremo di fornire un’ampia panoramica delle ammidi. Forniremo una breve panoramica della nomenclatura delle ammidi, due importanti proprietà delle ammidi che differiscono notevolmente dalle ammine, e andremo oltre tre strategie chiave per la sintesi delle ammidi.
Tabella dei contenuti
- Nomenclatura del gruppo funzionale delle ammidi: Amidi primarie, secondarie e terziarie
- Amidi contro ammine: Meno basico, più acido
- Sintesi delle ammidi, parte 1. Sostituzione acilica nucleofila di alogenuri acilici (o anidridi) con ammine
- Sintesi delle ammidi, parte 2: Idrolisi parziale dei nitrili
- Sintesi delle ammidi, parte 3: Uso di un reagente disidratante (come il DCC)
- Sommario: tre metodi efficaci per la sintesi delle ammidi
- Prendiamo brevemente in considerazione un quarto metodo, meno importante: Forza bruta
- Note
- Quiz Yourself!
- (Avanzato) Riferimenti e ulteriori letture
1. Nomenclatura del gruppo funzionale dell’ammide: Amidi primari, secondari e terziari
“Amidi” sono ciò che chiamiamo un’ammina che ha un singolo gruppo carbonile attaccato. Il gruppo funzionale ammide sta alle ammine come gli esteri stanno agli alcoli.
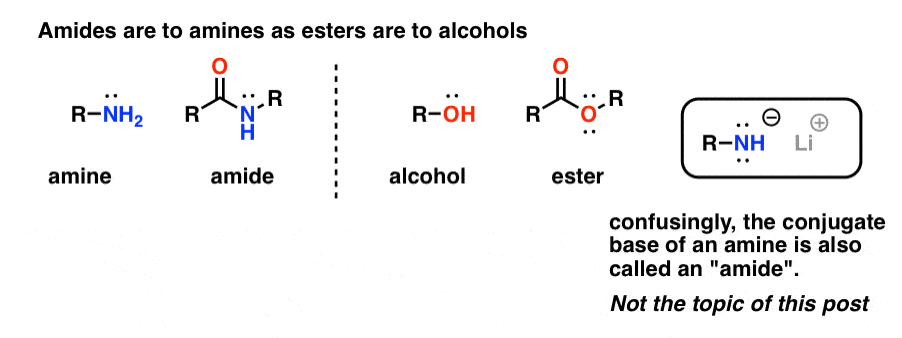
Confusamente, la parola “ammide” è usata anche per riferirsi alla base coniugata delle ammine, come l’ammide di sodio (NaNH2) e la di-isopropilamide di litio (LDA). Questi ultimi sono talvolta differenziati riferendosi a loro come “basi amidiche”. Altri useranno una pronuncia leggermente diversa per differenziarle (ayyy-myde e aaah-midd). Come per qualsiasi altro omonimo, la chiave è il contesto.
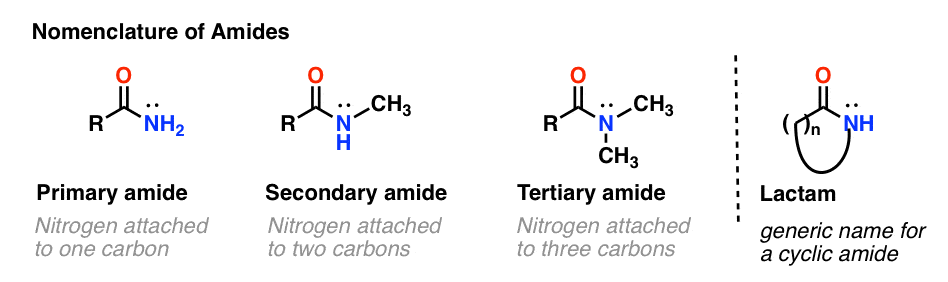
Come per le ammine, la nomenclatura usata per un’ammide dipende dal numero di carboni attaccati all’azoto.
Un’ammide primaria (1°) ha l’azoto attaccato a un solo carbonio; un’ammide secondaria (2°) ha l’azoto attaccato a due carboni; un’ammide terziaria (3°) ha l’azoto attaccato a tre carboni. Un’ammide ciclica è chiamata lattame.
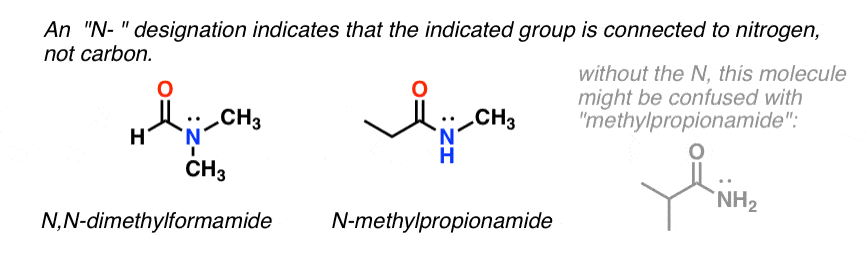
Quando l’azoto dell’ammide ha sostituenti diversi dall’idrogeno, li specifichiamo usando il prefisso N- per evitare confusione. Per esempio, N-metilpropionammide specifica l’attaccamento di un gruppo metilico sull’azoto; senza il prefisso N-, si potrebbe supporre che il gruppo metilico sia attaccato al carbonio, che sarebbe una molecola completamente diversa.
Amidi contro ammine: Meno basico, più acido
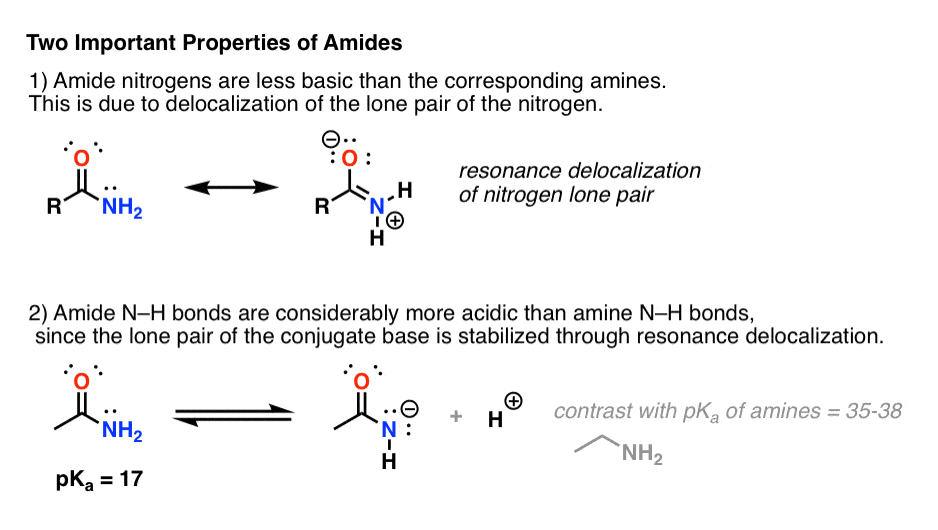
Attaccare un gruppo carbonilico a un’ammina ha due effetti drastici sulle proprietà dell’azoto.
- In primo luogo, i nitrogeni delle ammidi sono considerevolmente meno basici dei nitrogeni delle ammine. Questo è principalmente il risultato della delocalizzazione della coppia solitaria dell’azoto nel legame pi greco del carbonile. Infatti, la posizione più basica di un’ammide non è l’azoto ma l’ossigeno (!).
- In secondo luogo, i legami N-H delle ammidi sono molto più acidi dei legami N-H delle ammine. Perché? Ancora la delocalizzazione. Il gruppo carbonilico attaccato permette alla coppia solitaria della base coniugata di essere delocalizzata attraverso la risonanza. Il pKa dell’acetammide (17) è circa 20 ordini di grandezza più acido dell’ammoniaca (38).
Una terza, più sottile proprietà delle ammidi è che solitamente hanno una rotazione limitata intorno al legame C-N. La forma di risonanza dove c’è un legame C-N dà un contributo così significativo all’ibrido di risonanza che si può pensare che il legame C-N abbia un “carattere parziale di doppio legame”.
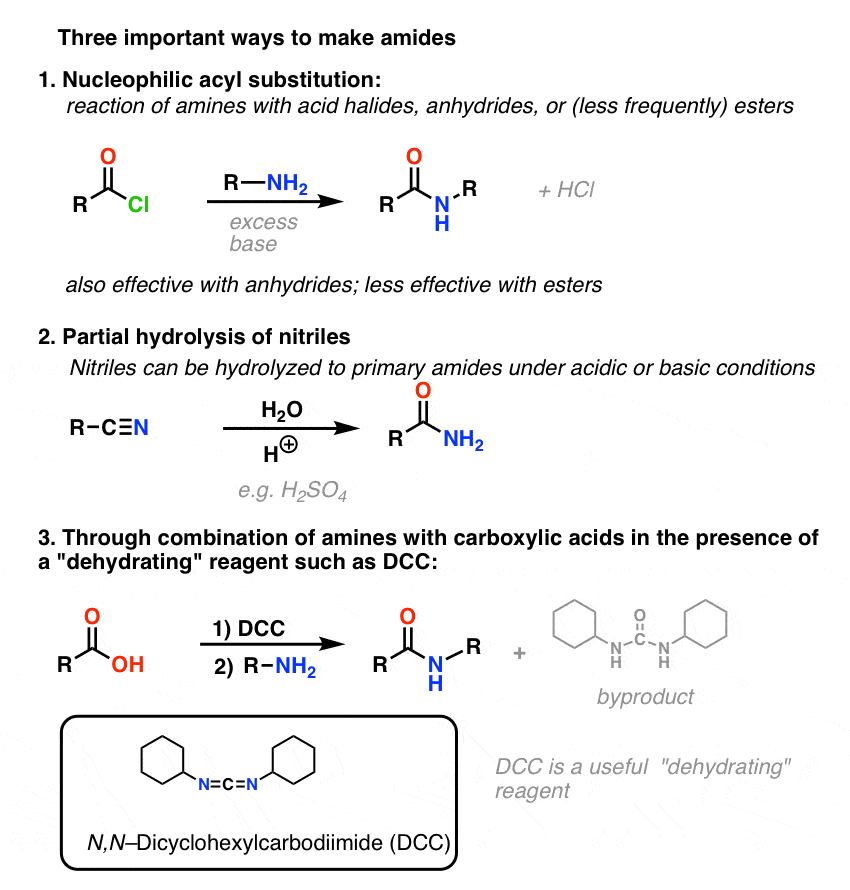
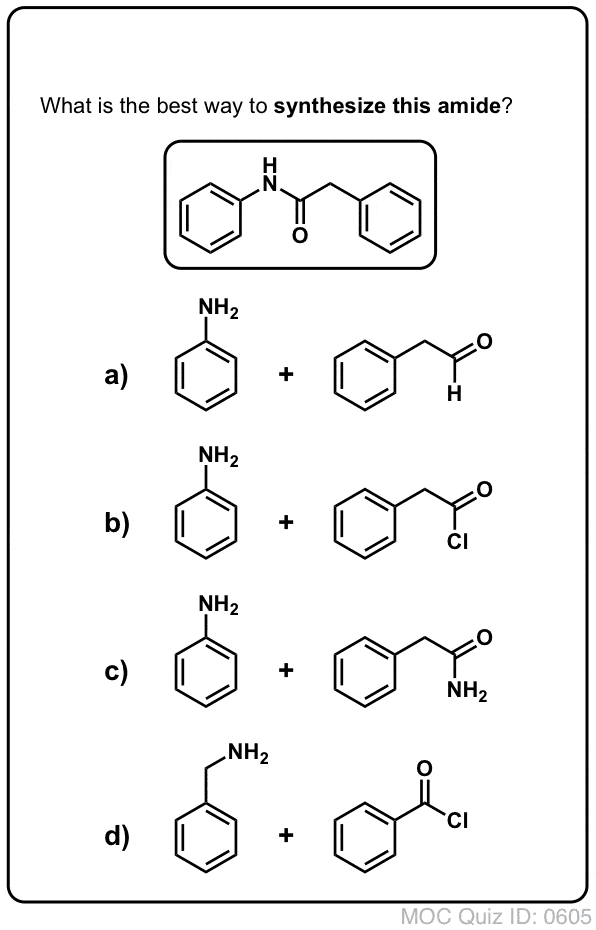
Sintesi delle ammidi, parte 1. Sostituzione acilica nucleofila di alogenuri acilici (o anidridi) con ammine
I gruppi acilici attaccati a un buon gruppo di partenza come cloruri acidi o anidridi acide possono facilmente subire una sostituzione acilica nucleofila con nucleofili ammine.
Se solo l’acido carbossilico è disponibile per iniziare, usare un reagente come il cloruro di tionile (SOCl2) per convertire un acido carbossilico in un cloruro acido è un buon primo passo per trasformare un acido carbossilico in un’ammide. (Anche PCl3, PCl5, cloruro di ossalile e una serie di altri reagenti possono funzionare). In alternativa, trattando un acido carbossilico con un alogenuro acilico si otterrà un’anidride, che può anche essere efficace.
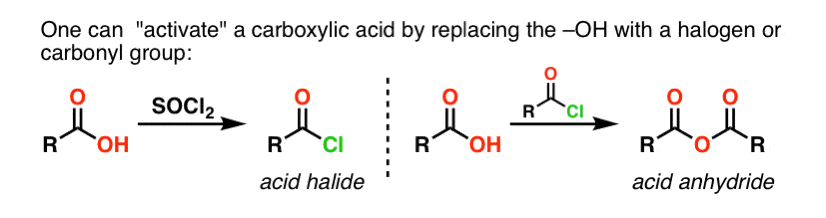
Gli alogenuri (ad esempio Cl- ) e i carbossilati (RCO2- ) sono basi molto più deboli, e quindi gruppi di partenza molto migliori di HO-. Quindi, aggiungendo un’ammina a un alogenuro acilico o a un’anidride acida, la sostituzione nucleofila acilica può avvenire in condizioni molto più blande, ottenendo la nostra ammide desiderata.
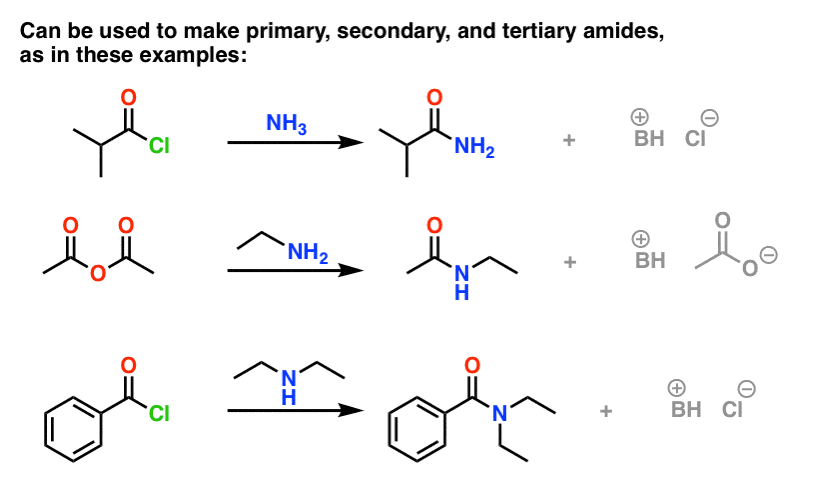
(Si possono ottenere ammidi attraverso la reazione di esteri con ammine, ma dato che gli alcossidi sono gruppi di partenza più poveri di alogenuri o carbossilati, questo metodo richiede condizioni più forzate. )
Una cosa da notare con gli alogenuri acidi è che il processo genera un singolo equivalente di HCl come sottoprodotto. In assenza di qualsiasi base aggiuntiva, la resa massima della procedura sarebbe del 50%, poiché l’HCl protonerebbe qualsiasi ammina e la renderebbe un sale di ammonio non nucleofilo.
Un modo per assicurare che la reazione proceda a completamento è aggiungere un secondo equivalente di ammina. Ci sono altri modi pratici per risolvere questo problema, che ho consegnato ad una nota a piè di pagina.
Se hai bisogno di un aggiornamento sul meccanismo della sostituzione nucleofila acilica passa il mouse qui per un’immagine pop-up o apri il link dell’immagine qui:
.
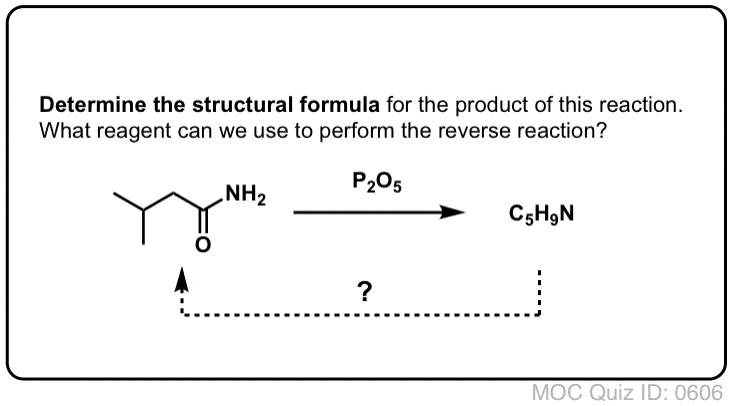
Sintesi delle ammidi, parte 2: idrolisi parziale dei nitrili
Un modo per pensare ai nitrili è che sono acidi carbossilici mascherati. Se trattati con acido acquoso e molto calore – condizioni da mazza – possono essere idrolizzati ad acidi carbossilici.
Uno degli intermedi in questo processo è un’ammide primaria.
Quindi se usiamo una tecnica di mazza leggermente più gentile e delicata, a volte è possibile salvare l’ammide dalla nostra miscela di reazione prima che sia idrolizzata ad acido carbossilico.
L’immagine qui sotto mostra come si potrebbe sintetizzare un’ammide da un precursore alogenuro alchilico, attraverso la reazione SN2 good-ol’:
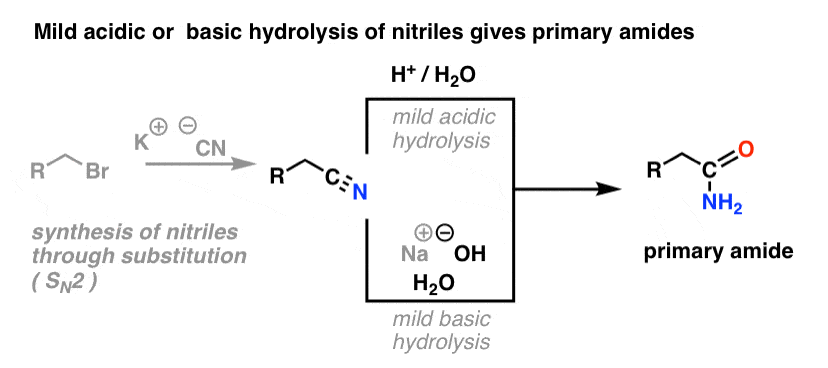
Cosa si intende per “blanda”? Una serie di condizioni per l’idrolisi della fenilacetammide (PhCH2CN) a PhCH2CONH2 dà le condizioni di reazione come “HCl, H2O, 40-50°C, 1h”.
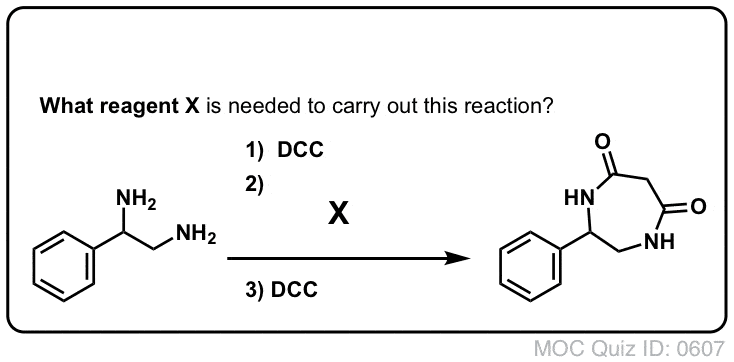
Sintesi delle ammidi, parte 3: uso di un reagente disidratante (come il DCC)
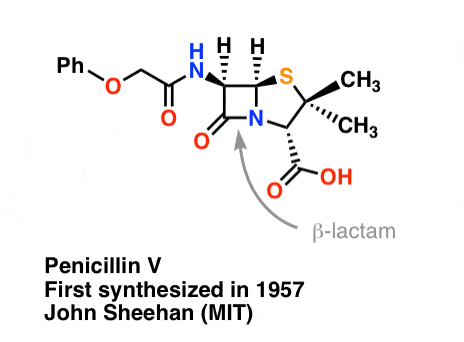
La sintesi della penicillina V nel 1957 dal gruppo di John Sheehan al MIT è uno dei risultati eroici della chimica organica del dopoguerra. Il problema chiave era la costruzione di un’ammide ciclica (l’anello β-lattamico) che è estremamente instabile in condizioni acide. Questo era di non poca importanza, poiché il β-lattame è anche la chiave del meccanismo d’azione della penicillina: interferire con la sintesi della parete cellulare batterica. I tentativi di fare questo ammide ciclico convertendo un acido carbossilico in un alogenuro acilico con SOCl2, PCl3, PCl5, e una miriade di altri metodi sono tutti falliti.
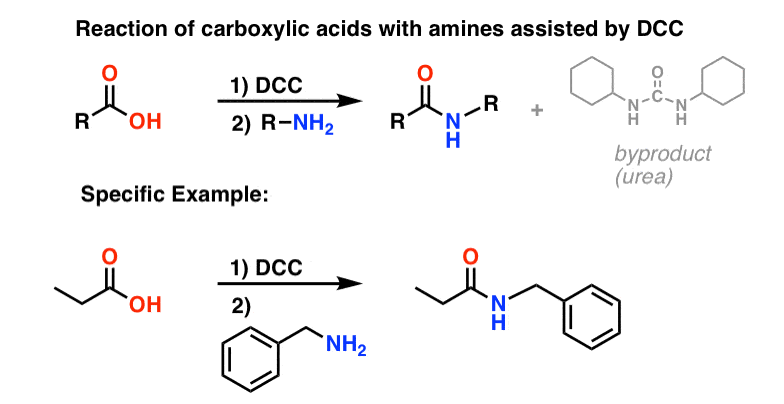
In risposta, il gruppo di Sheehan ha intelligentemente inventato un reagente disidratante molto delicato: N, N’-dicyclohexylcarbodiimide (DCC) che permetteva la formazione di ammidi in condizioni molto blande a pH neutro.
Oggi, il DCC (e il suo cugino più pratico (nota), EDC) sono ampiamente usati per la sintesi di ammidi sensibili – in particolare peptidi – in condizioni molto delicate.
Nelle condizioni di reazione, l’ossigeno carbossilato si attacca al carbonio elettrofilo del DCC, facendo quello che noi chiamiamo un “estere attivo” – in altre parole, un estere che ha effettivamente un gruppo di partenza decente (a differenza della maggior parte degli esteri, che non lo fanno). L’estere attivo viene poi attaccato dall’ammina in una classica sostituzione nucleofila acilica, che porta alla formazione dell’ammide.
Guardando il sottoprodotto, si nota che ci sono due idrogeni (ciascuno attaccato all’azoto) e un ossigeno (attaccato al carbonio centrale). Ecco dove è andato l’H2O!
Perché questo post sta diventando ridicolmente lungo, ho salvato il meccanismo per questa immagine pop-up.
Siccome questa reazione avviene in condizioni neutre, è estremamente utile nella sintesi dei peptidi, che possono subire racemizzazione (epimerizzazione, in realtà) in condizioni sia basiche che acide.
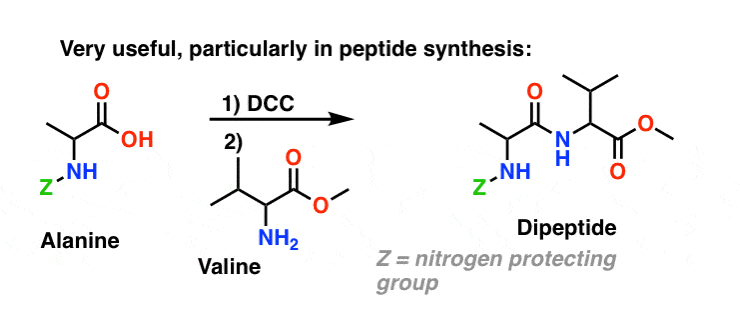
Sommario: tre metodi efficaci per la sintesi delle ammidi
Finiamo riassumendo questi tre importanti (ma per nulla esaustivi) modi per fare ammidi:
Questo conclude il nostro post sui punti principali della nomenclatura delle ammidi, proprietà e sintesi. Per un metodo bonus di sintesi delle ammidi, continuate a leggere.
Consideriamo brevemente un quarto metodo, meno importante: Forza bruta
Siccome di solito è coperto nei libri di testo, concludiamo considerando una quarta possibilità – la più semplice che si possa immaginare. Cosa succede se prendiamo un acido carbossilico e lo combiniamo con un’ammina, sperando che si formi un’ammide. Cosa succede?
Le ammine sono basi, e gli acidi carbossilici sono, beh, acidi. Aggiungi i due insieme e otterrai un innocuo sale.
A volte si possono fare ammidi riscaldando questo sale in un tubo sigillato, facendo uscire un equivalente di acqua. Questo metodo è chiamato pirolisi.
Il metodo non è niente se non diretto, e ha tutta la sottigliezza di un obice.
Il problema con la pirolisi è che il gruppo HO- di un acido carbossilico è un terribile gruppo di partenza.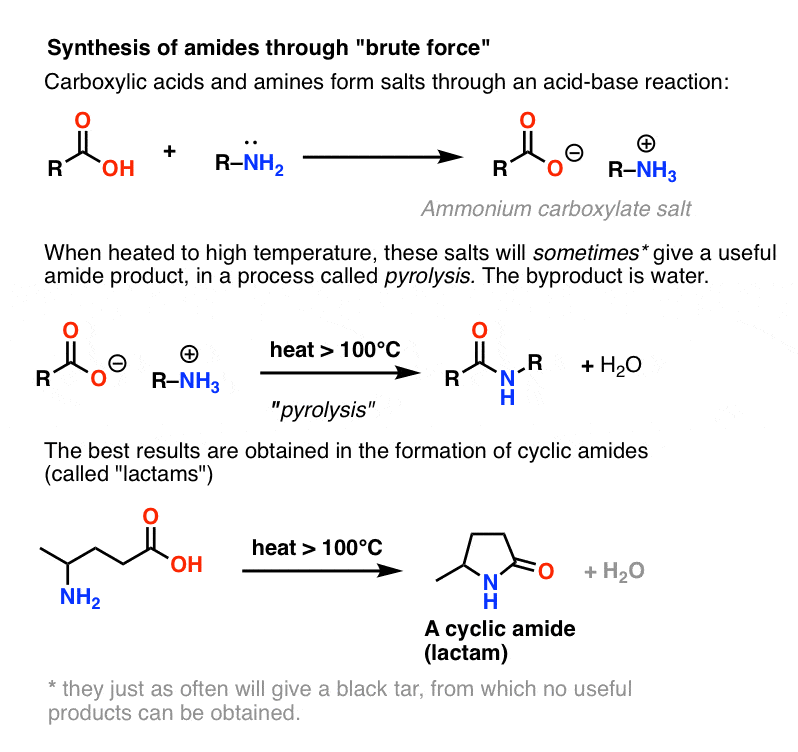
Per formare un’ammide da questa specie, l’ossigeno carbossilato (O- ) deve in qualche modo partire. Questa non è una cosa facile da fare, poiché la base coniugata di O- è il doppio anione O2- . In qualsiasi elenco di gruppi di partenza, O2- si collocherebbe da qualche parte tra “merda” e “f&maledettamente terribile”.
Tuttavia, se si colpisce questo sale con l’equivalente chimico del Martello di Thor: forza bruta, calore elevato, si può verificare una serie di trasferimenti di protoni dal sale di ammonio. alla fine si libera H2O e si forma il legame C-N.
Questo processo è chiamato pirolisi (pyro = fuoco, lysis = rottura).
In certi casi, specialmente ammidi semplici, e anche nella formazione di semplici lattami, il processo può essere soddisfacente.
In molti altri casi, tuttavia, risulta nella formazione di un catrame nero sul fondo del vostro pallone da cui non si può ottenere alcun prodotto utile.
Come ogni chimico organico può dirvi, ci sono molti modi diversi per creare catrame nero intrattabile sul fondo del vostro pallone, e questo è solo un metodo. Pensate a quante cose avete ancora da scoprire!

La ricerca per immagini su Google “Hammer of Thor” era legata alla chimica, ma sorprendentemente NSFW.
Note
Un articolo divertente e correlato: Ammidi: Humble But Useful (da Chemical & Engineering News).
C’è anche un effetto induttivo, per cui l’ossigeno elettronegativo (elettronegatività di 3,44) tira sugli elettroni del carbonio attaccato, che a sua volta tira sugli elettroni dell’azoto.
Nota 2. Un modo molto comune di realizzare questa reazione è quello di usare le cosiddette condizioni di Schotten-Baumann, dove si prendono i reagenti in un solvente come l’etere dietilico o il dicorometano, e si aggiunge una soluzione acquosa di NaOH, ottenendo una miscela bifasica. I sali di ammonio che si formano possono dissolversi nella fase acquosa, dopodiché vengono neutralizzati dalla base in eccesso e ritornano nella fase organica. Le ammine sono generalmente molto più nucleofile degli ioni idrossido, quindi l’idrolisi del cloruro acido per dare un acido carbossilico non è generalmente un problema.
Nota 3.
“Al momento della mia sintesi di successo della penicillina V nel 1957, ho paragonato il problema di cercare di sintetizzare la penicillina con metodi classici a quello di tentare di riparare la molla di un bell’orologio con incudine, martello e pinze di un fabbro” – John C. Sheehan
Nota 4. Il problema con l’uso del DCC è che il sottoprodotto, il DCU, è una tremenda rottura di palle da eliminare. La maggior parte dei sottoprodotti sono facilmente rimossi usando la cromatografia a colonna. Non il DCU. Prestando poca attenzione alla polarità del solvente, il DCU emerge da una colonna lentamente, a gocce e gocce, contaminando ogni frazione mentre va. L’EDC è una variante del DCC che ha un’unità di ammina terziaria; così, un semplice lavaggio acido durante la lavorazione rimuoverà tutta l’urea, risparmiando molto tempo e mal di testa.
L’immagine seguente mostra l’ultimo passo della sintesi di Sheehan usando il DCC.
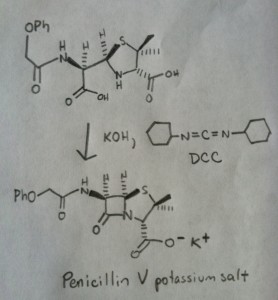
Imagine: Carmen Drahl/Chemical & Engineering News
Quiz Yourself!
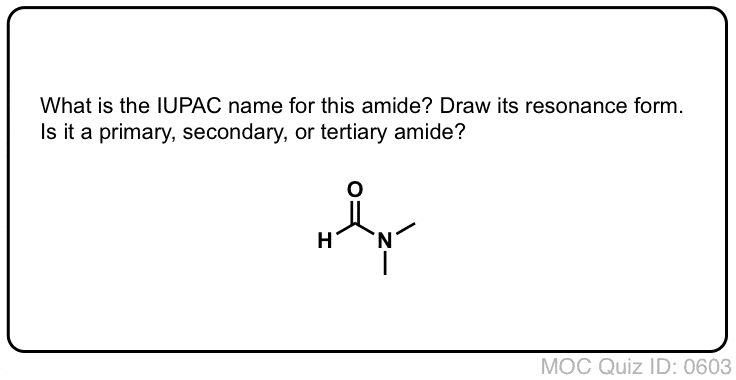 Clicca per lanciare
Clicca per lanciare 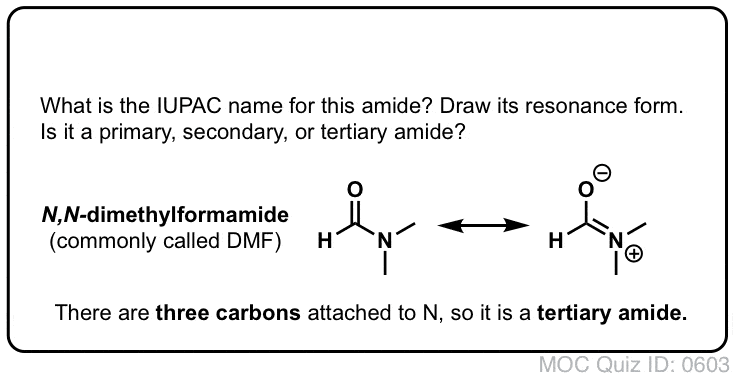
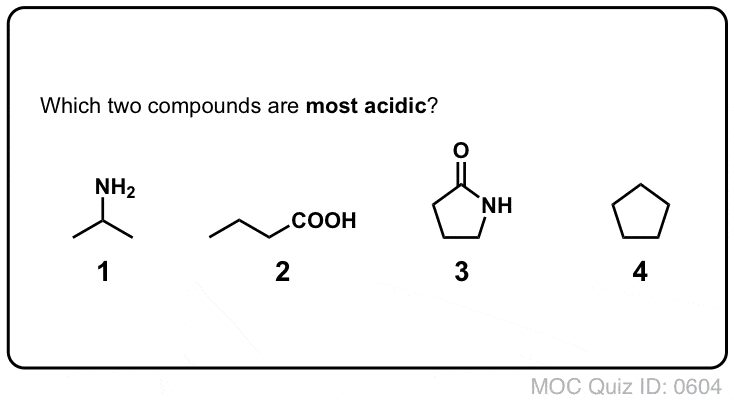 Clicca per girare
Clicca per girare 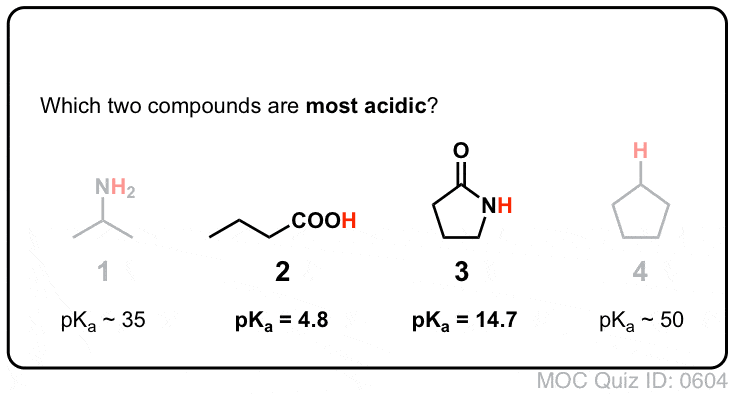
 Click to Flip
Click to Flip 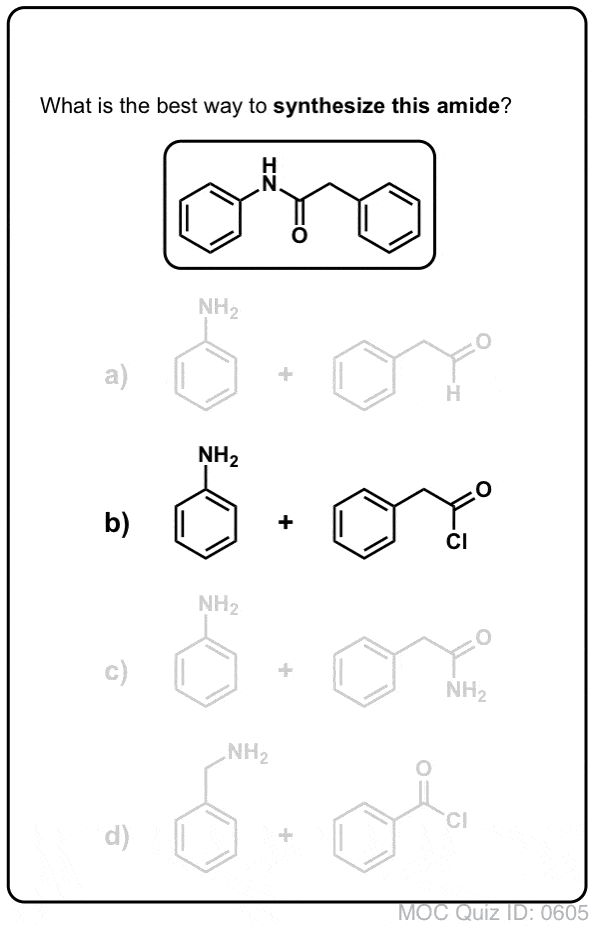
 Clicca per capovolgere
Clicca per capovolgere 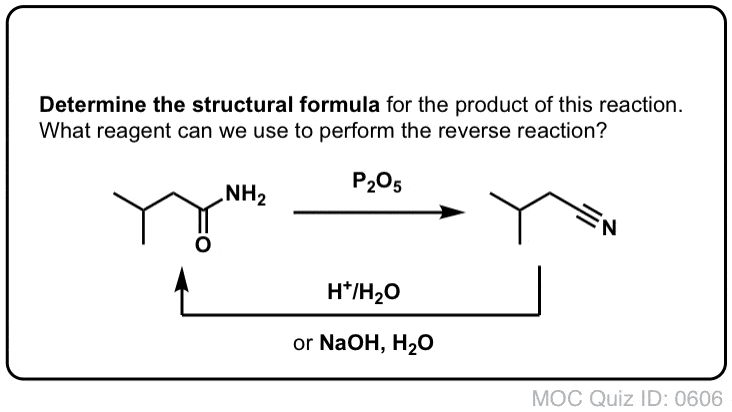
 Click to Flip
Click to Flip 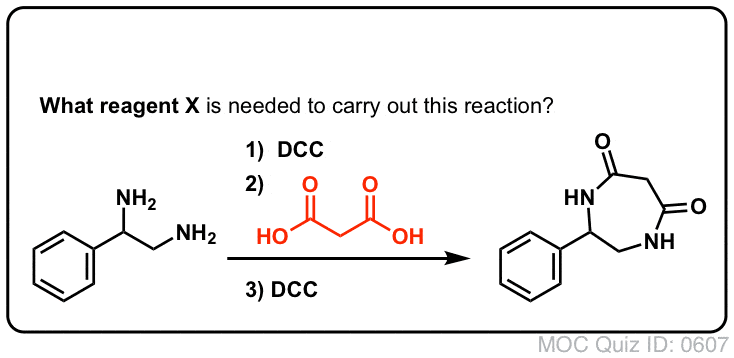
Riferimenti e ulteriori letture (avanzate)
L’idrolisi al nitrile:
- FENILACETAMIDE
Wilhelm Wenner
Org Synth. 1952, 32, 92
DOI: 10.15227/orgsyn.032.0092
Le condizioni usate qui per l’idratazione del nitrile all’ammide sono piuttosto dolci – questo usa una temperatura di 40 °C per circa 1 ora. - Idrolisi del nitrile guidata da alogenuri
James M. Photis
Tetrahedron Lett. 1980, 21 (37), 3539-3540
DOI: 10.1016/0040-4039(80)80228-0
Questa è una procedura utile per l’idrolisi selettiva dei nitrili in ammidi primarie, specialmente nel caso degli aroil cianuri (per esempio PhCOCN). - Conversione facile e altamente selettiva di nitrili ad ammidi tramite idratazione catalizzata acido indiretto utilizzando TFA o AcOH-H2SO4
Jarugu Narasimha Moorthy e Nidhi Singhal
The Journal of Organic Chemistry 2005, 70 (5), 1926-1929
DOI: 10.1021/jo048240a Reazione Schotten-Bauman:
#4 e #5 sono gli articoli originali di Schotten e Baumann su una semplice sintesi ammidica bifase. - Ueber die Oxydation des Piperidins
Schotten, C.
Ber. 1884, 17 (2), 2544-2547
DOI: 10.1002/cber.188401702178 - Ueber eine einfache Methode der Darstellung von Benzoësäureäthern
Baumann, E.
Ber. 1886, 19 (2), 3218-3222
DOI: 10.1002/cber.188601902348 - Sintesi totale enantioselettiva di (-)-Kibdelone C
John R. Butler, Chao Wang, Jianwei Bian, e Joseph M. Ready
Journal of the American Chemical Society 2011, 133 (26), 9956-9959
DOI: 1021/ja204040k
L’umile reazione di Schotten-Baumann è persino usata in sintesi totali impegnative – in questo caso, è usata per fare il lattame in 4 da 5 e 6! - BENZOYL PIPERIDINE
Marvel, C. S.; Lazier, W. A.
Org. Synth. 1929, 9, 16
DOI: 10.15227/orgsyn.009.0016
Questa procedura da Organic Syntheses, una fonte di procedure di laboratorio organico sintetico indipendentemente testate e riproducibili, è una classica sintesi ammidica di Schotten-Baumann. - Un processo ad alta produttività per il Valsartan
Ulrich Beutler, Matthias Boehm, Peter C. Fuenfschilling, Thomas Heinz, Jean-Paul Mutz, Ulrich Onken, Martin Mueller, and Werner Zaugg
Organic Process Research & Development 2007, 11 (5), 892-898
DOI: 1021/op700120n
Organic Process & Research Development (“OPRD”) è una grande rivista per la chimica di processo o scale-up. Questo articolo mostra come la reazione di Schotten-Baumann (da 4 a 3) sia preferita per reazioni su larga scala in quanto è semplice, robusta, facilmente realizzabile e non ha grandi esoterme (a differenza della reazione di Grignard, per esempio). - Il trucco della corda di nylon: Demonstration of condensation polymerization
Paul W. Morgan and Stephanie L. Kwolek
Journal of Chemical Education 1959, 36 (4), 182
DOI: 1021/ed036p182
Il classico ‘trucco della corda di nylon’ in cui si tira un filo di nylon da una miscela bifasica di esametilendiammina e sebacoyl chloride può essere considerato un tipo di reazione di Schotten-Baumann, in quanto forma una poliammide! Questo è stato sviluppato per la prima volta da Stephanie Kwolek, che è stata un’illustre chimica della DuPont per oltre 40 anni ed è stata responsabile della scoperta del Kevlar e dello sviluppo della chimica delle aramidi e di altri materiali ad alta resistenza: - Un nuovo metodo per formare legami peptidici
John C. Sheehan e George P. Hess
Journal of the American Chemical Society 1955, 77 (4), 1067-1068
DOI: 1021/ja01609a099
Uno studio originale sulla sintesi di legami peptidici/ammide utilizzando la DCC.DeTar ha pubblicato una serie di articoli che studiano il meccanismo delle reazioni di formazione dei legami mediate dal DCC e da altri carbodiimmidi, ed ecco i primi due: - Reazioni dei Carbodiimmidi. I. The Mechanisms of the Reactions of Acetic Acid with Dicyclohexylcarbodiimide
DeLos F. DeTar and Richard Silverstein
Journal of the American Chemical Society 1966, 88 (5), 1013-1019
DOI: 10.1021/ja00957a027 - Reactions of Carbodiimides. II. Le reazioni della dicloesilcarbodiimmide con acidi carbossilici in presenza di ammine e fenoli
DeLos F. DeTar e Richard Silverstein
Journal of the American Chemical Society 1966, 88 (5), 1020-1023
DOI:1021/ja00957a028 - The Chemistry of Carbodiimides.
G. Khorana
Chemical Reviews 1953, 53 (2), 145-166
DOI: 10.1021/cr60165a001
Una vecchia recensione del Prof. Har Gobind Khorana, che più tardi ricevette il premio Nobel per la medicina per il suo lavoro che dimostra che i nucleotidi nel DNA e RNA codificano per la sintesi proteica. - ESTERIFICAZIONE DI ACIDI CARBOXILICI CON DICICLOEOSSICARBODIIMIDE/4-DIMETHYLAMINOPYRIDINE: tert-BUTYL ETHYL FUMARATE
Neises e Wolfgang Steglich
Org. Synth. 1985, 63, 183
DOI: 10.15227/orgsyn.063.0183
Questa è una procedura per l’esterificazione selettiva usando DCC – questo evita la transesterificazione che avverrebbe nelle condizioni usuali di esterificazione di Fischer. Questa procedura è da Organic Syntheses, una fonte di reazioni organiche sintetiche affidabili e testate indipendentemente. - Sintesi peptidica in fase solida. I. La sintesi di un tetrapeptide
B. Merrifield
Journal of the American Chemical Society 1963, 85 (14), 2149-2154
DOI: 10.1021/ja00897a025
Questo è uno degli articoli più citati in JACS, e per una buona ragione – fondamentalmente pone le basi della SPPS, e quello che ora è un settore da miliardi di dollari. Questo lavoro ha portato a un premio Nobel per la chimica per l’autore, il Prof. R. Bruce Merrifield (Rockefeller U.). Gli accoppiamenti peptidici sono fatti usando nientemeno che il DCC. - Note- Una sintesi conveniente di Carbodiimidi solubili in acqua.
John Sheehan, Philip Cruickshank, e Gregory Boshart
The Journal of Organic Chemistry 1961, 26 (7), 2525-2528
DOI:1021/jo01351a600
Lo svantaggio principale con DCC è che la separazione del DCU (dicicloesilurea) così prodotto può essere ingombrante. Così sono stati sviluppati altri reagenti, come l’EDC (1-Ethyl-3-(3′-dimethylaminopropyl)carbodiimide), per il quale l’urea risultante è solubile in acqua e facilmente rimossa per estrazione. - Sintesi totale di un antibiotico monociclico peptide lattone, etamicina
John C. Sheehan and Stephen L. Ledis
Journal of the American Chemical Society 1973, 95 (3), 875-879
DOI:1021/ja00784a041
EDC è stato usato per la maggior parte degli accoppiamenti peptidici nella sintesi di questo peptide, che è uno dei primi peptidi ciclici ad essere prodotto sinteticamente.